Te hiciste tu perfil metabólico y todo salió bien. Pero sigues subiendo de peso con facilidad, te cuesta bajar esas lonjitas tercas del abdomen, te mueres de sueño después de comer y la energía de las 4pm ya no regresa. ¿Y si el problema no es el estrés, sino una resistencia a la insulina silenciosa que tus análisis todavía no detectan?
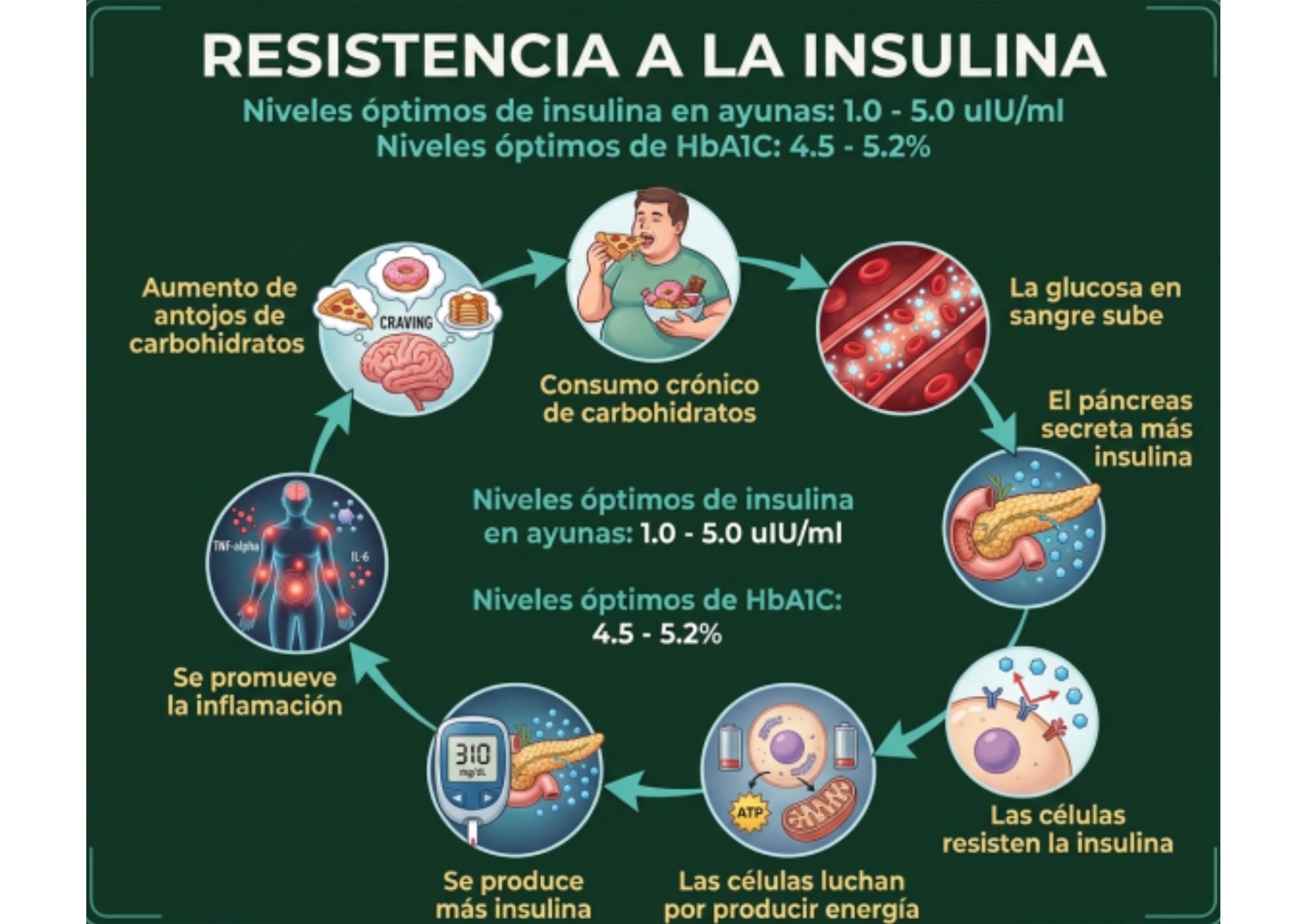
Durante años, la medicina convencional ha tratado la resistencia a la insulina (RI) como un fenómeno binario: o tienes diabetes tipo 2, o no la tienes. Si tu glucosa en ayunas sale en rango y tu HOMA-IR no se dispara, te dicen que estás “bien”. El problema es que la ciencia molecular de la última década —incluyendo revisiones recientes publicadas en Nature Reviews Molecular Cell Biology y en Signal Transduction and Targeted Therapy— describe algo muy distinto: la resistencia a la insulina no aparece de un día para otro. Es un proceso que comienza en el tejido adiposo años antes de que cualquier análisis de sangre convencional se altere.
Esto no es un matiz académico. Es la diferencia entre intervenir a tiempo o esperar a que el daño metabólico ya esté instalado.
En este artículo vamos a revisar qué dice la ciencia molecular más reciente sobre cómo se desarrolla la resistencia a la insulina, por qué el HOMA-IR —la prueba que casi todo el mundo usa para “descartarla”— llega sistemáticamente tarde, qué papel puede jugar la bioimpedancia como herramienta de detección temprana, y qué cambios de estilo de vida y alimentación tienen, hoy, el respaldo más sólido para revertir el proceso antes de que se convierta en diagnóstico.
1. Lo que sentías no era exageración: el cuerpo cambia mucho antes de que el laboratorio lo confirme
Empecemos por validar algo que escuchamos todos los días en consulta: “mis análisis salen bien, pero yo no me siento bien.”
Esa frase no es un signo de hipocondría ni de ansiedad sin fundamento. Es, literalmente, lo que predice la fisiología de la resistencia a la insulina. El cuerpo no envía una sola señal de alarma cuando algo empieza a fallar; envía docenas de señales pequeñas —fatiga después de comer, antojo de carbohidratos a media tarde, dificultad para bajar grasa abdominal, niebla mental, sueño no reparador— mucho antes de que la bioquímica clásica (glucosa e insulina en ayunas) se mueva lo suficiente para disparar una alarma en un reporte de laboratorio.
La razón tiene una explicación molecular concreta, y vale la pena entenderla, porque cambia por completo cómo deberíamos vigilar nuestra salud metabólica.
2. Qué es —y qué no es— la resistencia a la insulina, según la ciencia actual
La definición clásica dice que la resistencia a la insulina es un estado en el que se necesita más insulina de la habitual para lograr el mismo efecto sobre la glucosa. Es correcta, pero incompleta. La revisión de James, Stöckli y Birnbaum en Nature Reviews Molecular Cell Biology (2021) propone algo más preciso: la resistencia a la insulina es, ante todo, un defecto en la capacidad de las vesículas de almacenamiento del transportador GLUT4 para trasladarse hacia la superficie de las células musculares y adiposas cuando la insulina lo ordena. Es decir, el problema no está, en la mayoría de los casos, en que falten receptores de insulina o en que la insulina “no funcione” de forma genérica. El problema está en un paso muy específico y muy distal de la cascada de señalización: la translocación de GLUT4.
Esto importa clínicamente porque desmonta un mito que sigue circulando en redes y en consultorios: que la resistencia a la insulina es simplemente “comer mucha azúcar” o “tener sobrepeso”. La obesidad y el exceso de azúcar son factores de riesgo importantes, sí, pero no son la causa molecular en sí misma. De hecho, existen personas delgadas con resistencia a la insulina significativa —los familiares de primer grado de pacientes con diabetes tipo 2 son el ejemplo clásico estudiado en la literatura— y personas con obesidad que mantienen una sensibilidad a la insulina notablemente conservada (lo que se conoce como “obesidad metabólicamente sana”). El peso en la báscula, por sí solo, nunca ha sido un buen proxy de lo que está pasando a nivel celular.
El verdadero protagonista: el tejido adiposo que ya no puede almacenar grasa de forma segura
Una de las actualizaciones conceptuales más importantes de los últimos años es esta: la resistencia a la insulina probablemente no empieza en el músculo ni en el hígado. Empieza en el tejido adiposo, cuando este pierde la capacidad de expandirse de forma sana.
El tejido adiposo puede crecer de dos formas:
- Hiperplasia: se generan nuevas células grasas (adipocitos) a partir de precursores. Es la forma “sana” de expansión.
- Hipertrofia: las células grasas existentes se inflan de tamaño porque no se están generando células nuevas que repartan la carga.
Cuando el tejido adiposo no logra expandirse por hiperplasia —ya sea por factores genéticos, inflamación o senescencia celular— y se ve forzado a crecer por hipertrofia, ocurre algo crítico: esas células grasas hipertrofiadas pierden capacidad de almacenamiento, reclutan macrófagos proinflamatorios, reducen su secreción de adiponectina (una de las pocas hormonas verdaderamente protectoras que conocemos en metabolismo) y empiezan a “derramar” ácidos grasos libres hacia la circulación. Ese derrame de lípidos hacia órganos que no están diseñados para almacenarlos —músculo, hígado, páncreas— es lo que se conoce como lipotoxicidad, y es uno de los motores centrales de la resistencia a la insulina sistémica.
Lo interesante —y lo que conecta directamente con la necesidad de una detección más temprana— es que estudios genéticos en humanos (incluyendo análisis de más de 50 loci asociados a resistencia a la insulina) muestran que buena parte del riesgo genético no está en los genes del músculo o del hígado, sino en genes que regulan la diferenciación y expansión del tejido adiposo. Esto sugiere que la “decisión” biológica de cómo va a crecer tu tejido graso —de forma sana o de forma tóxica— se empieza a jugar mucho antes de que aparezca cualquier alteración detectable en sangre.
Ceramidas, diacilgliceroles y el estrés mitocondrial: el lenguaje molecular del daño
Cuando el exceso de ácidos grasos libres llega a células que no son adipocitos, no se queda ahí sin consecuencias. Se transforma en moléculas con efectos directos sobre la señalización de insulina, principalmente:
- Ceramidas: lípidos que se acumulan en músculo, hígado e incluso en mitocondrias, y que se asocian de forma consistente con resistencia a la insulina en estudios tanto humanos como animales. Las ceramidas de cadena larga (16 y 18 carbonos) son las más implicadas. Interfieren con la activación de AKT —una pieza clave de la cascada de señalización de la insulina— y promueven fragmentación mitocondrial y producción de especies reactivas de oxígeno (ROS).
- Diacilgliceroles (DAG): activan isoformas específicas de la proteína quinasa C (PKC-θ en músculo, PKC-ε en hígado) que terminan fosforilando e inhibiendo al receptor de insulina o a sus sustratos inmediatos.
- Disfunción mitocondrial y bajos niveles de coenzima Q: trabajos recientes muestran que niveles reducidos de coenzima Q en la mitocondria generan estrés reductivo y aumentan la producción de especies reactivas de oxígeno, contribuyendo de forma directa a la resistencia a la insulina, independientemente de cuánta grasa total tenga una persona.
Aquí hay un punto que vale la pena subrayar porque cambia la forma de explicarle esto a un paciente: la evidencia actual sugiere que los defectos “proximales” de la señalización de insulina —es decir, fallas en el receptor de insulina o en las primeras moléculas de la cascada (IRS, PI3K, AKT)— tienen mucha “redundancia” biológica. El sistema está diseñado con un margen de seguridad tan amplio que reducir de forma moderada la actividad de estas moléculas no suele traducirse en resistencia a la insulina clínicamente relevante. El verdadero cuello de botella está más adelante, en los componentes distales de la cascada —sobre todo en la translocación de GLUT4— y es ahí donde convergen el estrés mitocondrial, las ceramidas y los DAG.
El concepto de “resistencia a la insulina selectiva”: por qué el hígado no resiste a todo por igual
Otro hallazgo que ha cambiado la forma de entender esto: en el hígado, la resistencia a la insulina no es uniforme. La insulina pierde la capacidad de frenar la producción de glucosa hepática (gluconeogénesis), pero mantiene intacta, e incluso exagera, su capacidad de estimular la producción de grasa nueva (lipogénesis de novo). A este fenómeno se le llama resistencia a la insulina selectiva, y explica por qué muchas personas con resistencia a la insulina desarrollan hígado graso (ahora llamado MAFLD, enfermedad hepática asociada a disfunción metabólica) incluso antes de mostrar alteraciones claras en su glucosa.
Esto tiene una implicación práctica enorme: la combinación de glucosa elevada y producción de grasa hepática elevada al mismo tiempo —en lugar de cancelarse— se retroalimenta. Más insulina circulando para compensar la resistencia hepática a la glucosa significa, al mismo tiempo, más estímulo para fabricar grasa en el hígado. Es un círculo que se autoalimenta silenciosamente.
El papel de la hiperinsulinemia: ¿causa o consecuencia?
Un debate que sigue abierto en la literatura, y que vale la pena mencionar porque es intelectualmente honesto reconocerlo: no sabemos con certeza absoluta si el exceso de insulina circulante (hiperinsulinemia) es la causa original de la resistencia a la insulina, o si es una consecuencia compensatoria que, una vez instalada, termina empeorando el problema. Lo que sí sabemos es que ambas coexisten casi siempre, y que la hiperinsulinemia crónica por sí sola —independientemente de su origen— puede degradar componentes de la señalización de insulina, promover inflamación en el tejido adiposo y favorecer aumento de peso. Es decir: panorama de círculo vicioso, no de causa única y lineal.
El papel silencioso de la epigenética y los microRNAs
Hay otra capa de la biología que la medicina convencional rara vez menciona en consulta, y que ayuda a explicar por qué dos personas con el mismo peso y el mismo estilo de vida pueden tener trayectorias metabólicas completamente distintas: la regulación epigenética.
La metilación del ADN en genes clave de la señalización de insulina —el propio gen de la insulina, el receptor de insulina, IRS1, los genes del IGF1— cambia de forma medible en presencia de sobrenutrición y obesidad, y esos cambios pueden alterar la expresión de estos genes años antes de que se traduzcan en un cuadro clínico reconocible. De forma similar, microRNAs específicos —pequeñas moléculas de ARN no codificante— regulan directamente la expresión de GLUT4, la función de las células beta del páncreas y el metabolismo de lípidos en el hígado. Algunos de estos microRNAs ya se están explorando como biomarcadores circulantes capaces de señalar alteraciones metabólicas antes de que aparezcan clínicamente.
¿Por qué importa esto para alguien que no es bioquímico? Porque refuerza la idea central de este artículo: el cuerpo “sabe” que algo está cambiando a nivel molecular mucho antes de que ese cambio se traduzca en un número alterado en un papel de laboratorio convencional. La pregunta no es si existe una ventana de detección temprana —la evidencia molecular confirma que sí existe—, la pregunta es qué herramientas tenemos disponibles, hoy, para asomarnos a esa ventana.
Lo que pasa en el intestino no se queda en el intestino
Un hallazgo que ha ganado fuerza en los últimos años, y que conecta directamente con algo que en medicina funcional trabajamos de forma rutinaria: la microbiota intestinal participa de forma activa en el desarrollo de la resistencia a la insulina. Cuando la glucosa elevada y la inflamación de bajo grado alteran la permeabilidad del intestino, fragmentos bacterianos como el lipopolisacárido (LPS) logran pasar hacia la circulación sistémica. Ese LPS circulante activa vías inflamatorias en el hígado y el tejido adiposo que empeoran directamente la resistencia a la insulina. Es un círculo que conecta intestino, inflamación y metabolismo de la glucosa de una forma que la medicina fragmentada por especialidades —un gastroenterólogo por un lado, un endocrinólogo por otro, sin que nadie vea el cuadro completo— difícilmente puede abordar de forma integrada.
3. Por qué el HOMA-IR llega tarde (y por qué esto no es un defecto de la prueba, sino de su diseño)
El HOMA-IR (Homeostatic Model Assessment of Insulin Resistance) sigue siendo una herramienta útil y ampliamente validada. El problema no es que esté “mal calculado”; el problema es qué tan tarde en el proceso biológico se vuelve anormal.
El HOMA-IR se basa en dos valores: glucosa e insulina en ayunas. Para que el HOMA-IR se eleve de forma clínicamente significativa, el cuerpo necesita estar ya secretando cantidades notablemente mayores de insulina solo para mantener la glucosa en rango. Es decir, el HOMA-IR detecta el momento en el que el páncreas ya está compensando de forma evidente. No detecta el momento en el que el tejido adiposo empezó a expandirse de forma hipertrófica, ni el momento en el que las primeras ceramidas comenzaron a acumularse en el músculo, ni el momento en el que la adiponectina empezó a caer.
Dicho de otra forma: el HOMA-IR mide el resultado tardío de un proceso que, según la evidencia de estudios de seguimiento a largo plazo en cohortes humanas, puede estar gestándose durante años en forma de cambios en la composición corporal —específicamente en la distribución y comportamiento del tejido graso— antes de traducirse en una alteración detectable de glucosa e insulina en ayunas.
Esto no es una opinión aislada. Estudios de seguimiento longitudinal en distintas poblaciones han documentado que los cambios en grasa visceral —medida por tomografía o por absorciometría— preceden, y predicen de forma independiente, el deterioro futuro de la sensibilidad a la insulina medida por HOMA-IR a 10 años de distancia. En otras cohortes, la acumulación progresiva de grasa visceral y la redistribución de grasa corporal predijeron el desarrollo de intolerancia a la glucosa y diabetes tipo 2 hasta 13 años antes del diagnóstico clínico. El patrón se repite: el tejido adiposo manda señales de alarma —en su tamaño, en su distribución, en su comportamiento celular— mucho antes de que el eje glucosa-insulina en ayunas se altere lo suficiente para que un HOMA-IR salga positivo.
Esta es la pregunta que debería estarse haciendo cualquier persona con antecedentes familiares de diabetes, síndrome metabólico o resistencia a la insulina: si el HOMA-IR detecta el problema cuando ya lleva años desarrollándose, ¿qué herramienta puede ayudarme a ver lo que está pasando antes de que el páncreas tenga que compensar?
4. El papel de la bioimpedancia: por qué la composición corporal habla antes que el laboratorio
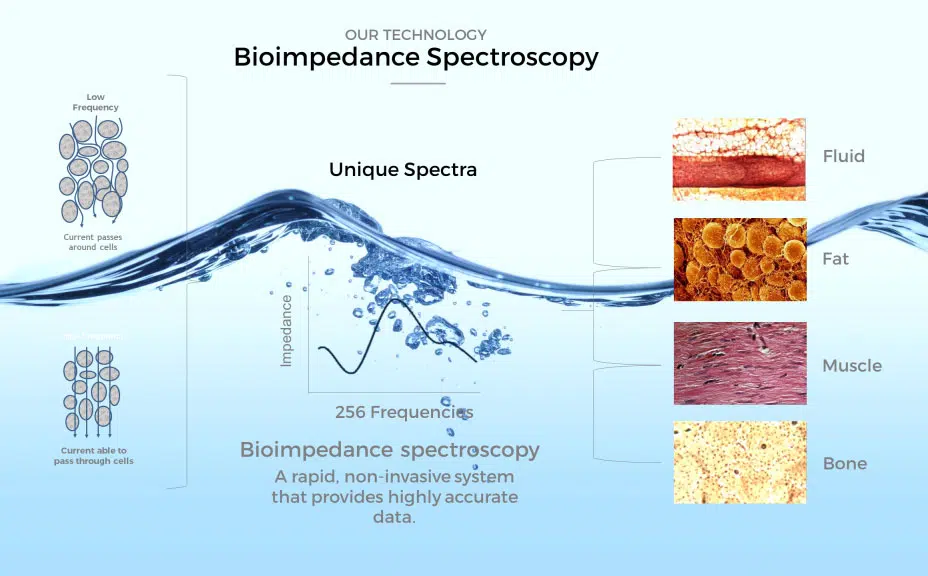
Aquí es donde entra una herramienta que en medicina funcional usamos de forma sistemática, y que en la medicina convencional sigue siendo profundamente subutilizada: el análisis de bioimpedancia (BIA).
La bioimpedancia no mide glucosa ni insulina. Mide cómo se distribuye una corriente eléctrica de bajo voltaje a través de los distintos tejidos del cuerpo, y a partir de esa información estima con buena precisión clínica:
- Grasa visceral: el tipo de grasa que, según la evidencia revisada arriba, es el predictor más temprano y más fuerte de deterioro futuro de la sensibilidad a la insulina —más fuerte, de hecho, que el índice de masa corporal o que la circunferencia de cintura por sí solos.
- Masa muscular y su distribución: el músculo es el principal sitio de eliminación de glucosa estimulada por insulina en el cuerpo; perder masa muscular de forma silenciosa (algo que ocurre años antes de notarse en el espejo) reduce directamente la capacidad del cuerpo para manejar glucosa.
- Ángulo de fase: un parámetro derivado de la bioimpedancia que refleja, a nivel celular, la integridad de las membranas y la salud general de la célula. Estudios recientes en pacientes con diabetes tipo 2 han encontrado una correlación inversa entre el ángulo de fase y el HOMA-IR: a menor ángulo de fase, mayor resistencia a la insulina. Y este parámetro empieza a deteriorarse antes de que el cuadro metabólico sea evidente en sangre, porque refleja cambios celulares —no solo bioquímicos— que ocurren de forma progresiva.
Lo que la evidencia de cohortes longitudinales sugiere —y lo que en la práctica clínica observamos de forma consistente— es que la trayectoria de la grasa visceral y del ángulo de fase a lo largo del tiempo puede anticipar, en algunas personas, el deterioro de la sensibilidad a la insulina medido por HOMA-IR por una década o más. No porque la bioimpedancia “vea el futuro”, sino porque está midiendo directamente el órgano (el tejido adiposo) donde, según la ciencia molecular que revisamos arriba, comienza realmente el proceso. El HOMA-IR mide la consecuencia río abajo; la bioimpedancia —usada de forma seriada, no como una sola fotografía— permite observar el cambio en el terreno río arriba.
Esto no significa que la bioimpedancia sustituya al laboratorio. Significa que es una pieza que, combinada con biomarcadores específicos (no solo glucosa e insulina en ayunas, sino ferritina, perfil tiroideo completo con T3 libre, relación triglicéridos/HDL, e insulina en ayunas interpretada con criterios funcionales y no solo convencionales), permite construir una fotografía mucho más temprana y mucho más completa de hacia dónde va tu metabolismo.
La pregunta clínicamente relevante deja de ser “¿tengo o no tengo resistencia a la insulina?” y se convierte en “¿hacia dónde se está moviendo mi composición corporal en los últimos 12 a 24 meses?” Esa es una pregunta que el HOMA-IR, por diseño, no puede responder, y que la bioimpedancia seriada sí puede empezar a contestar.
Por qué una sola medición de bioimpedancia no basta
Vale la pena ser precisos en algo que con frecuencia se simplifica de más: un solo estudio de bioimpedancia, tomado de forma aislada, tiene el mismo problema que una sola foto de glucosa en ayunas: te dice dónde estás hoy, pero no te dice hacia dónde te diriges. El valor real de esta herramienta está en su uso seriado —idealmente cada 6 meses, en condiciones similares de hidratación, horario y actividad física previa— porque lo que predice el deterioro futuro de la sensibilidad a la insulina no es un número absoluto de grasa visceral, sino su tendencia a lo largo del tiempo.
Esto cambia la conversación clínica de una forma importante. En lugar de decirle a una persona “tu grasa visceral está en rango normal, no te preocupes”, la pregunta correcta es: “¿tu grasa visceral subió, bajó o se mantuvo estable en los últimos seis meses, y qué cambió en tu vida durante ese periodo?” Esa segunda pregunta es la que realmente conecta con la fisiología que describimos en las secciones anteriores —hipertrofia adipocitaria progresiva, caída de adiponectina, derrame de lípidos hacia músculo e hígado— y la que permite intervenir mientras el proceso todavía es reversible con cambios de estilo de vida, antes de que se vuelva necesario un manejo farmacológico.
Qué buscamos específicamente en un reporte de bioimpedancia clínica
No todos los parámetros que arroja un estudio de bioimpedancia tienen el mismo valor predictivo. En la práctica clínica funcional, los que vigilamos con más atención cuando el objetivo es detección temprana de resistencia a la insulina son:
- Grasa visceral en valor absoluto y en tendencia, porque es, de los índices de obesidad disponibles, el que muestra mayor capacidad predictiva sobre la resistencia a la insulina del tejido adiposo en estudios comparativos frente a índice de masa corporal, circunferencia de cintura y porcentaje de grasa corporal total.
- Ángulo de fase, porque refleja integridad celular y se correlaciona de forma inversa con el HOMA-IR, especialmente en hombres, lo que lo convierte en un marcador complementario útil cuando el cuadro bioquímico todavía no es concluyente.
- Masa muscular esquelética en relación con el peso total, porque la pérdida silenciosa de masa muscular —común después de los 35-40 años si no hay entrenamiento de fuerza— reduce directamente la capacidad del cuerpo para disponer de glucosa, incluso si el peso en la báscula no cambia.
- Agua intracelular vs. extracelular, porque cambios en esta relación pueden reflejar inflamación sistémica de bajo grado, un componente activo del proceso que lleva a la resistencia a la insulina.
Ningún parámetro de bioimpedancia, por sí solo, diagnostica resistencia a la insulina —y es importante decirlo con esa claridad, porque la transparencia sobre los límites de cada herramienta es lo que distingue a la medicina funcional rigurosa de la medicina funcional que solo vende tecnología impresionante sin contexto clínico—. Lo que aporta es una ventana hacia el órgano donde, según la evidencia molecular revisada en este artículo, el proceso probablemente comienza.
5. Entonces, ¿cómo se ve la detección temprana en la práctica?
Proponemos un modelo de vigilancia metabólica con tres capas, pensado para detectar el proceso años antes de que se vuelva un diagnóstico:
Capa 1 — Composición corporal (cada 6 a 12 meses)
Bioimpedancia con seguimiento de grasa visceral, masa muscular y ángulo de fase. Lo que importa no es el valor absoluto de una sola medición, sino la tendencia en el tiempo.
Capa 2 — Bioquímica funcional (anual, o antes si hay síntomas)
Insulina y glucosa en ayunas (sí, siguen siendo útiles, solo que no son suficientes por sí solas), hemoglobina glucosilada, perfil lipídico completo con cálculo de la relación triglicéridos/HDL (un proxy barato y subutilizado de resistencia a la insulina), ferritina, y perfil tiroideo completo incluyendo T3 libre —porque la disfunción tiroidea subclínica y la resistencia a la insulina con frecuencia coexisten y se potencian mutuamente.
Capa 3 — Historia clínica funcional
Las preguntas que casi nunca se hacen en una consulta de 10 minutos: ¿hay antecedentes familiares de diabetes tipo 2? ¿Cómo cambió tu energía después de los 35-40 años? ¿Tienes antojo de carbohidratos en horarios predecibles? ¿Tu sueño es reparador? ¿Cuánto tiempo llevas con grasa abdominal que no responde a dieta ni ejercicio convencional?
Ninguna de estas tres capas, por separado, da el panorama completo. Juntas, permiten ver el proceso metabólico años antes de que cumpla los criterios diagnósticos convencionales de resistencia a la insulina o prediabetes.
6. Lo que sí cambia el rumbo: alimentación y estilo de vida con respaldo real
Aquí es donde la conversación se pone honesta: no existe ningún medicamento aprobado específicamente para “tratar” la resistencia a la insulina como entidad propia. Lo que existe —y lo que tiene la evidencia más sólida y consistente, por encima de cualquier suplemento de moda— son cambios de estilo de vida. Vamos a revisarlos con el mismo rigor con el que revisamos la biología arriba, sin convertir esto en una lista genérica de “come más verduras”.
6.1 Movimiento: no todo el ejercicio actúa igual sobre la insulina
El ejercicio mejora la sensibilidad a la insulina por una vía que es independiente de la insulina misma: la contracción muscular activa la AMPK, que estimula la translocación de GLUT4 hacia la superficie celular sin necesitar que la insulina haga ese trabajo. Esto es clínicamente importante porque significa que el músculo en movimiento puede “saltarse” parte del problema de resistencia a la insulina, captando glucosa por una puerta alterna.
Lo que la evidencia respalda con más fuerza:
- Entrenamiento de resistencia (fuerza): aumenta la masa muscular, que es el principal tejido de disposición de glucosa del cuerpo. Más masa muscular metabólicamente activa significa más “espacio de almacenamiento” para la glucosa circulante. Estudios en modelos de dieta alta en grasa muestran que programas estructurados de entrenamiento de resistencia reducen el peso, el índice de obesidad, el área de los adipocitos y la inflamación crónica de bajo grado, mejorando la resistencia a la insulina de forma medible.
- Ejercicio aeróbico regular: incrementa la biogénesis mitocondrial en el músculo a través de la activación de PGC1-alpha, lo cual mejora directamente la capacidad oxidativa y reduce la acumulación de lípidos tóxicos intramusculares.
- La combinación, no la dosis heroica, es lo que funciona: 30 minutos de actividad física, al menos cinco días a la semana, activan AMPK de forma suficiente para mejorar la translocación de GLUT4 y aumentar la captación de glucosa. No se necesita entrenar como atleta de alto rendimiento; se necesita constancia.
- Lo que el sedentarismo hace en sentido contrario: estudios de reposo en cama de apenas 7 a 9 días muestran reducción medible de la acción de la insulina en el músculo. Esto significa que la inactividad —no solo el sobrepeso— es, por sí misma, un generador activo de resistencia a la insulina, incluso en personas delgadas.
6.2 Alimentación: menos sobre restricción y más sobre la señal metabólica que envías
Las recomendaciones nutricionales con mejor respaldo no dependen de un único “villano” (no es solo el azúcar, no es solo la grasa); dependen de cómo cada patrón de alimentación afecta la carga lipídica, la inflamación y la sensibilidad celular a la insulina.
Reducir ultraprocesados y azúcares de absorción rápida, no por moralismo, sino por mecanismo.
El consumo elevado de carbohidratos refinados genera picos repetidos de insulina que, sostenidos en el tiempo, contribuyen a la hiperinsulinemia compensatoria. Esto no significa eliminar carbohidratos por completo —las dietas muy bajas en carbohidratos sostenidas a largo plazo tienen su propio perfil de riesgo— sino priorizar carbohidratos de absorción lenta, con fibra, y evitar el patrón de “pico-caída” que perpetúa el antojo de las 4pm que tantas pacientes describen.
Priorizar fibra y patrón de plato, no conteo obsesivo.
La evidencia respalda de forma consistente patrones de alimentación ricos en fibra soluble, vegetales de hoja, legumbres y proteína magra como estrategia para mejorar la sensibilidad a la insulina, en parte porque modulan la velocidad de absorción de glucosa y en parte porque favorecen una microbiota intestinal asociada a menor inflamación sistémica.
El eje intestino-metabolismo no es opcional, es central.
La evidencia actual muestra que la disbiosis intestinal —alteración en la diversidad y composición de la microbiota— se asocia de forma consistente con mayor riesgo de resistencia a la insulina, en parte a través de la producción alterada de ácidos grasos de cadena corta y del aumento de permeabilidad intestinal, que permite el paso de lipopolisacáridos bacterianos hacia la circulación, generando inflamación crónica de bajo grado en hígado y tejido adiposo. Esto conecta directamente con algo que en medicina funcional llevamos años trabajando de forma integrada: la salud intestinal y la sensibilidad a la insulina no son dos temas separados, son la misma conversación.
Grasas: calidad e contexto, no cantidad ciega.
La sobrealimentación con grasa, especialmente en contextos de exceso calórico sostenido, induce resistencia a la insulina hepática y muscular de forma medible incluso en personas sanas. Pero no toda grasa actúa igual: los ácidos grasos saturados en exceso generan más ceramidas y DAG que los ácidos grasos insaturados, lo cual respalda priorizar fuentes de grasa como pescado, aceite de oliva, frutos secos y aguacate por encima de grasas saturadas en exceso.
Ayuno intermitente: una herramienta, no una solución universal.
El ayuno intermitente puede mejorar marcadores de sensibilidad a la insulina en algunas personas al reducir la frecuencia de picos de insulina y favorecer la oxidación de grasa durante las ventanas de ayuno. Pero su efecto depende enormemente del contexto individual —nivel de estrés, calidad de sueño, historial de relación con la comida— y no debe prescribirse de forma genérica sin valorar a la persona completa.
6.3 Sueño y ritmo circadiano: el factor que casi nunca se menciona en una consulta de 10 minutos
La privación de sueño y la disrupción del ritmo circadiano —turnos rotativos, jet lag social, exposición a luz artificial en la noche— se asocian de forma creciente con alteraciones en la composición y función de la microbiota intestinal, y con deterioro medible de la sensibilidad a la insulina, independientemente del peso corporal. Dormir mal no es un síntoma colateral de la resistencia a la insulina: es, en sí mismo, un mecanismo que la perpetúa.
6.4 Estrés crónico: el cortisol no es un concepto de bienestar, es un mecanismo bioquímico
El cortisol elevado de forma sostenida interfiere directamente con la supresión de la producción hepática de glucosa y con la utilización periférica de glucosa por el músculo. Esto explica por qué el estrés crónico —no el estrés puntual de un examen o una entrega, sino el estrés sostenido durante meses o años, tan característico de la vida profesional en una ciudad como la Ciudad de México— tiene un correlato bioquímico medible sobre la resistencia a la insulina, y por qué cualquier estrategia de intervención que ignore el manejo del estrés está, por diseño, incompleta.
6.5 Pérdida de peso moderada: la magnitud que sí importa
Un hallazgo consistente en la literatura: perder entre el 5% y el 7% del peso corporal —no el 20%, no una transformación estética completa— es suficiente para reducir significativamente el riesgo de progresión hacia diabetes tipo 2 y para mejorar de forma medible la sensibilidad a la insulina en personas con sobrepeso u obesidad. Esto es clínicamente liberador, porque desmonta la idea de que solo una pérdida de peso drástica genera beneficio metabólico real.
7. Lo que la medicina convencional no está mal en hacer, pero sí en hacer sola
Vale la pena ser honestos aquí, porque la honestidad radical es parte de cómo entendemos la medicina funcional: no decimos que la medicina convencional esté equivocada en usar HOMA-IR, glucosa en ayunas o hemoglobina glucosilada. Estas herramientas son válidas, están bien estudiadas y siguen siendo el estándar para el diagnóstico formal de resistencia a la insulina y diabetes tipo 2.
Lo que decimos es que, usadas solas, llegan sistemáticamente tarde respecto al proceso biológico real. Y que existe evidencia robusta —la que revisamos arriba sobre grasa visceral, ángulo de fase, ceramidas y disfunción mitocondrial— que permite construir un panorama mucho más temprano, sin necesidad de esperar a que el páncreas ya esté compensando de forma evidente.
Esa es la diferencia entre medicina reactiva y medicina preventiva real: no es magia, no es “intuición”, es seguir la cascada molecular hasta su origen y vigilar ahí, en lugar de esperar a que la consecuencia final se vuelva visible en un papel.
Cómo lo trabajamos en Sanar
Desde 2004 hemos visto, consulta tras consulta, el mismo patrón: pacientes con años de síntomas metabólicos y laboratorios “normales”, que finalmente reciben un diagnóstico cuando el proceso ya lleva mucho tiempo en marcha. Por eso nuestro protocolo de valoración metabólica combina bioimpedancia seriada, perfil bioquímico funcional —no solo los marcadores convencionales, sino los que la evidencia muestra que se alteran de forma más temprana— e historia clínica detallada, integrando intestino, tiroides y manejo del estrés como parte de la misma conversación, no como especialidades separadas. No prometemos resultados garantizados ni un protocolo único para todos; cada plan se ajusta según hacia dónde muestre que se está moviendo tu propio metabolismo.
7.5 Lo que vemos diferente en pacientes de la Ciudad de México
Hay un factor que casi ningún artículo sobre resistencia a la insulina menciona, y que en Polanco vemos todos los días en consulta: vivir y trabajar en la Ciudad de México no es un contexto metabólico neutro.
La altitud de la CDMX (alrededor de 2,240 metros) obliga al cuerpo a adaptarse a una menor disponibilidad de oxígeno, lo cual modifica la producción de glóbulos rojos y puede alterar marcadores como la ferritina y la saturación de hierro, complicando la interpretación de un perfil metabólico si no se ajusta el criterio clínico al contexto de altitud. La exposición sostenida a contaminación atmosférica se asocia, en la literatura, con mayor estrés oxidativo sistémico —el mismo tipo de estrés mitocondrial que revisamos arriba como motor de la resistencia a la insulina—. Y el patrón de vida del profesional urbano en esta ciudad —tráfico prolongado como fuente de estrés crónico de baja intensidad pero sostenido, jornadas laborales largas, cenas tardías, sueño fragmentado por el ruido y la actividad nocturna de una ciudad que no se detiene— combina casi todos los factores de riesgo modificables que describimos en este artículo: estrés crónico, mal sueño, inactividad y un patrón de alimentación frecuentemente reactivo en lugar de planeado.
Esto no es un detalle anecdótico: significa que, para una persona que vive y trabaja en CDMX, la ventana de detección temprana que describimos arriba probablemente debería abrirse antes, no después, que para alguien en un contexto de menor exposición a estos factores combinados.
7.6 Mitos frecuentes que retrasan el diagnóstico temprano
“Si no tengo sobrepeso, no puedo tener resistencia a la insulina.”
Falso. Como revisamos arriba, existen personas delgadas —particularmente familiares de primer grado de pacientes con diabetes tipo 2— con resistencia a la insulina medible en músculo e hígado, sin obesidad ni intolerancia a la glucosa evidente. El peso es un factor de riesgo, no un requisito.
“Mis análisis de glucosa siempre salen perfectos, entonces estoy descartada.”
La glucosa en ayunas es, de las pruebas convencionales, una de las que se altera más tarde en el proceso, precisamente porque el páncreas compensa produciendo más insulina durante años antes de “rendirse”. Una glucosa normal con una insulina elevada, o con una relación triglicéridos/HDL desfavorable, ya es una señal de alerta que merece atención.
“La resistencia a la insulina es lo mismo que la prediabetes.”
No exactamente. La prediabetes es un punto en el continuo —definido por criterios específicos de glucosa— que típicamente aparece después de años de resistencia a la insulina progresiva. Esperar a la prediabetes para actuar es, de nuevo, intervenir en una etapa tardía del proceso.
“Si tomo un suplemento de moda, lo resuelvo.”
Ningún suplemento sustituye la combinación de movimiento, alimentación, sueño y manejo del estrés que describimos en este artículo. Algunos micronutrientes muestran efectos modestos en estudios controlados, pero ninguno revierte por sí solo un proceso que involucra al tejido adiposo, al músculo, al hígado, al intestino y a la mitocondria de forma simultánea.
7.7 Condiciones que conviene vigilar en paralelo
La resistencia a la insulina rara vez viaja sola. La evidencia la asocia de forma consistente con disfunción tiroidea subclínica (donde una T3 libre baja en rango “normal” convencional puede coexistir con resistencia a la insulina sin que ningún perfil tiroideo básico lo detecte), con el síndrome de ovario poliquístico —donde la resistencia a la insulina y el exceso de andrógenos se retroalimentan en un círculo bidireccional—, con el hígado graso asociado a disfunción metabólica, y con un perfil inflamatorio de bajo grado que, sostenido durante años, incrementa el riesgo cardiovascular incluso antes de que aparezca un diagnóstico formal de diabetes. Por eso un abordaje funcional nunca evalúa la resistencia a la insulina de forma aislada: la pregunta siempre es cómo se conecta con el resto del sistema.
8. La pregunta que vale la pena hacerte
Si tus análisis de glucosa e insulina en ayunas siempre han salido “normales”, pero llevas años notando que tu cuerpo ya no responde como antes —que la grasa abdominal no se mueve, que la energía de media tarde no regresa, que el antojo de carbohidratos llega siempre a la misma hora— probablemente no estás imaginando nada. Es probable que tu tejido adiposo esté en medio de una transición que el laboratorio convencional todavía no puede ver.
¿Hace cuánto tiempo sientes que tu cuerpo ya no responde igual, aunque tus análisis sigan saliendo “bien”?
Cuéntanoslo en los comentarios. Y si quieres saber en qué punto de esa trayectoria está tu propio metabolismo —antes de que un análisis convencional te lo confirme— puedes escribirnos directamente por WhatsApp para agendar una valoración con bioimpedancia y perfil metabólico funcional.




